Soni Savai Pullamsetti, et al. Hypoxia-inducible factor signaling in pulmonary hypertension. The Journal of Clinical Investigation 2020


肺动脉高压(PH)以肺动脉重塑为特征,可最终导致右心衰竭和早期死亡。新的证据表明,缺氧诱导因子(HIF)在PH的发病机制中起着基础性和关键性的作用。本综述总结了HIF亚型的调节及其在各种PH亚型中的影响,以及揭示这一通路作用的详细条件性和细胞特异性基因敲除小鼠的研究。我们还讨论了HIF泛型和亚型选择性抑制剂的临床前研究现状,并提出了新的研究领域,以促进将HIF亚型特异性抑制作为一种新的治疗PH和右心衰的策略。


转录因子缺氧诱导因子(HIF)是氧稳态的主要调节因子,其作为异二聚体复合物,由氧敏感α亚基(HIF-α;包括HIF-1α、HIF-2α[EPAS1]和HIF-3α)和氧不敏感β亚基(HIF-β;包括HIF-1β [芳基烃受体核转运体,ARNT1]、ARNT2和ARNT3)组成。在氧化细胞中,HIF-α亚基通过脯氨酰羟化酶结构域蛋白(PHD)和HIF抑制因子(FIH)的羟基化作用而失活,从而结合von Hippel-Lindau(VHL)肿瘤抑制蛋白,该蛋白为E3泛素连接酶复合物的组成部分,随后靶定羟基化的HIF-α并进行蛋白酶体降解。在缺氧条件下,氧气变得有限,导致HIF-α羟基化减弱,从而使HIF-α亚基稳定。这启动了核移位以及HIF-α亚基和HIF-β亚基的结合。这种激活的HIF通过诱导或抑制大量参与调节血管张力、血管生成、红细胞生成、细胞代谢、增殖、存活和自噬的基因来启动对缺氧的适应性反应。然而,在非缺氧条件下,生长因子、激素或细胞因子也在不同水平上调节HIF-α亚基,并调节许多信号通路。在肺中,HIF协调对缺氧的生理反应,并参与多种疾病的发病机制,包括肺癌、慢性阻塞性肺疾病(COPD)、肺纤维化(PF),以及肺动脉高压。
肺动脉高压(PH)是一种严重的肺血管疾病,其特征是血管细胞过度增殖,细胞外基质沉积增加,肺血管壁内炎性细胞积聚,共同导致肺血管阻力增加。尽管在这一领域进行了广泛的研究,但疾病发展和进展的机制尚不完全清楚。在许多失调的信号通路中,HIF信号通路已被确定为决定疾病进展的一种潜在机制,不仅在肺动脉高压(PAH;I组PH)中,而且在肺疾病和/或缺氧引起的PH(包括与慢性高原暴露、COPD和PF(III组PH)相关的PH)中也发挥作用。值得注意的是,在PAH、慢性血栓栓塞性PH和特发性PF相关PH患者的肺组织中观察到HIF-1α表达增强,而HIF-2α与先天性膈疝相关PH相关。患有急性呼吸系统疾病相关PH的新生儿患者的循环HIF-1α水平也升高。此外,在紫绀和持续性PH的新生儿外周血细胞中,HIF-1α及其靶基因——血管内皮生长因子(VEGF)和促红细胞生成素(EPO)上调,因此HIF-1α是代表全身性缺氧的早期标志物。同样,在肺动脉高压患者中观察到的循环骨髓源性祖细胞的增加,受肺动脉内皮细胞(PAEC)中HIF-1α驱动的CXCL12(C-X-C基序趋化因子配体12)表达的调节。
PH患者肺组织HIF-1α表达增加的细胞来源是PAECs和肺动脉平滑肌细胞(PASMCs)。虽然一些报告显示肺动脉高压患者肺动脉中HIF-1α上调,但也有证据表明从特发性肺动脉高压(IPAH)患者分离的PASMC中HIF-1α降低的证据。PAH患者的肺组织中HIF-1β的表达也增加,同样,PAH和IPF相关PH患者的肺动脉中HIF-2α的表达也增加。PH患者肺组织中HIF-2α表达增加的细胞来源主要是PAEC,提示PH中存在细胞类型和环境特异性对HIF亚型的调节。
各种亚型PH中HIF的激活表明,除慢性缺氧外,还有其他导致PH发生的因素(基因变异、血管收缩、内皮功能障碍、线粒体异常、细胞生长失调和炎症)可以激活HIF信号通路,触发肺血管细胞、炎症细胞和心肌细胞的改变,从而重塑肺血管系统和右心室(RV)(图1)。

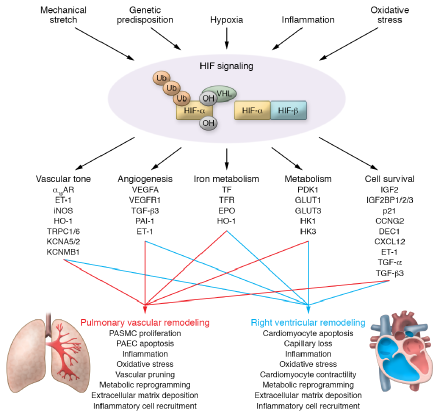
肺动脉高压中HIF信号转导的新概念。新的证据表明,除缺氧外,许多促PH因子(如炎症、机械牵张、氧化应激和遗传易感性)均聚集于HIF信号通路,导致血管张力、血管生成、代谢和细胞存活发生变化,随后导致肺血管和右心室重塑。
HIF通路的基因变异
在高海拔人群和Chuvash红细胞增多症患者中发现的HIF通路分子的基因变异说明了HIF通路在肺血管适应和重塑中的重要性。Chuvash红细胞增多症的特征是VHL中存在R200W(598C>T)错义突变,该突变降低其与羟基化HIF-α亚基的结合,从而增加HIF-1α和HIF-2α水平。这导致HIF靶基因的表达,包括EPO和VEGF,并导致红细胞增多症的发展。在Chuvash红细胞增多症中,除了红细胞增多症外,还发现一些VHL功能缺失突变,包括D126N(376G>A)、D126N(376G>A)/S183L(548C>T)和M54I(162G>C),其与较高的静息肺动脉压(PAP)、严重PH和RV功能障碍有关。此外,HIF-2αG537R的突变可损害HIF-2α羟基化,导致家族性红细胞增多症,也与PH有关。
对包括藏族人、埃塞俄比亚人和安第斯山脉人在内的高海拔人群进行了几项全基因组选择研究,并确定了HIF通路中及其周围基因突变的正向选择信号(使这些人群能够适应高海拔缺氧的生活)。然而,长期居住在高海拔地区可能会导致PAP的持续增加和PH的发展。在所有高海拔人口中,藏族人的PAP最低。一项基于全基因组分析结果的候选基因研究发现,EPAS1(HIF-2α)变异与藏族人的低PAP有关,该研究确定了与高原适应相关的基因变异。此外,生活在低海拔地区但携带EPAS1(编码HIF-2α)和EGLN1(编码PHD2)基因变异的藏族人表现出缺氧肺血管收缩减弱。因此,未来的研究需要阐明HIF通路基因变异在驱动当地高海拔人群对高海拔PH的敏感性或抗性方面的作用。
血管调节因子
各种血管调节因子已被证明可调节PAEC中HIF亚型的稳定性和转录活性(图2和表1)。一氧化氮(NO)维持肺血管张力,其下调与PH发病机制有关。最近的研究表明,NO在低氧/HIF轴调节中起着核心作用。例如,在PAEC中,缺氧导致顺式天然反义RNA sONE对内皮NO合酶(eNOS)表达的转录后负调控,从而导致NO水平降低和HIF-1α稳定性增加。因此,由于eNOS缺失导致的低水平NO也会稳定HIF-1α并导致常氧内皮细胞(ECs)的迁移。此外,内源性NO生成调节因子,如精氨酸酶-2和不对称二甲基精氨酸,通过稳定PAEC中的HIF-1α来影响细胞增殖和炎症基因表达。内皮素1(ET-1)是一种有效的血管收缩剂,也能稳定HIF-1α,进而通过eNOS介导的活性氧(ROS)的产生,促进HIF-1α诱导的糖酵解转换。同样,ET-1刺激常氧PASMC可增加HIF-1α的稳定性,这是由于增加了Ca2+和ROS。并由于ERK1/2通路激活而增加了HIF-1α的转录活性。ERK1/2途径激活使p300磷酸化以增加其与HIF-1α的结合。此外,ET-1通过钙调神经磷酸酶依赖的RACK1去磷酸化,促进常氧PASMC中HIF-1α蛋白的稳定,进而抑制PHD2活性。重要的是,HIF也被证明可以调节ET-1的合成。NEPAS(HIF-3α的转录变体)整体缺乏的小鼠表现出ET-1过表达,这导致肺血管重塑和扩张型心肌病,这是由于从出生开始就很明显的过度的组织血管化,并在生命的后期阶段发展。因此,HIF和ET-1形成一个双向调节回路,在驱动肺血管重塑中发挥重要作用。
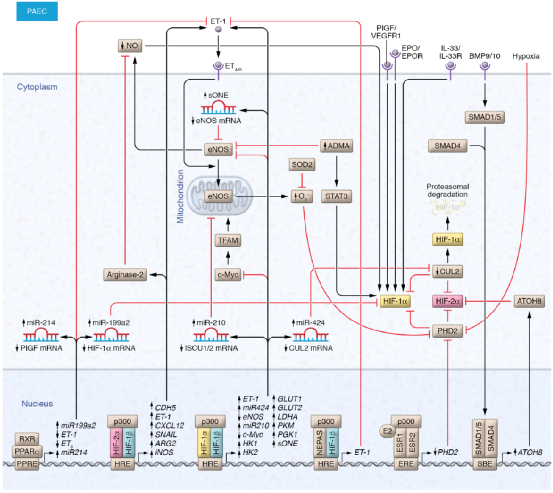
HIF信号在PH中的作用:肺动脉内皮细胞的上游和下游调节因子。在肺动脉内皮细胞(PAECs)中,血管调节、线粒体和炎症生长因子以及与PH相关的表观遗传异常调节HIF亚型稳定性和转录活性。随后,HIF亚型转录激活了一系列参与血管张力、血管生成、代谢和细胞增殖的基因。
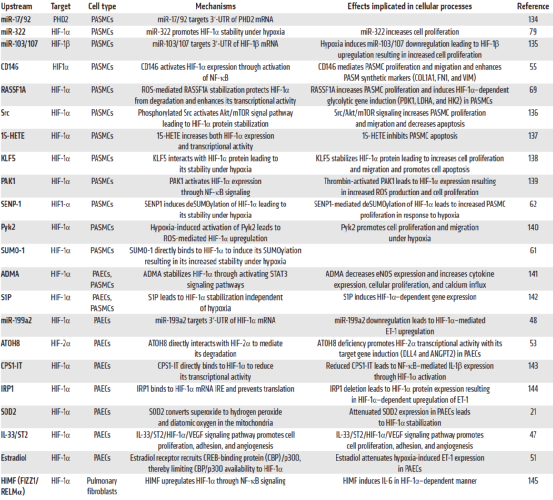
PH中HIF信号通路上游调控因子的总结
炎症、生长因子和micro RNA
缺氧诱导PAEC中IL-33及其受体ST2的上调,激活下游HIF-1α/VEGF信号,以ST2依赖的方式增强增殖、粘附和血管生成。类似地,激素和生长因子,如骨形态发生蛋白(BMP)、胎盘生长因子(PlGF)、血小板衍生生长因子(PDGF)、促红细胞生成素(EPO)、雌二醇和信号调节分子,可调节PAEC中的HIF亚型的转录活性。例如,在镰状细胞病相关(SCD相关)PH患者中,PlGF水平升高导致microRNA-199a2(miR-199a2)下调,microRNA-199a2是HIF-1α的负调节因子。此外,PPARα激动剂介导的miR-199a2转录减弱ET-1表达和HIF-1α水平,改善SCD小鼠模型的PH。雌二醇通过干扰HIF活性,从而负向调控ET-1在PAEC中的表达,可能是通过竞争性限制CBP/p300的数量。在缺氧PAEC中,雌二醇还通过促进雌激素受体β介导(ERβ介导)的PHD2上调,促进HIF-2α降解,从而负向调节HIF-2α。相比之下,BMP信号分子SMAD1和SMAD5转录激活bHLH转录因子8(ATOH8)表达,后者与HIF-2α相互作用,降低其在缺氧暴露的PAECs中的丰度及其靶基因δ样蛋白4 (DLL4)和血管生成素2 (ANGPT2)的表达。ATOH8-KO小鼠自发产生PH,表明BMP信号在调控HIF通路中起重要作用。此外,在人内皮细胞中,HIF-1α驱动的miR-322/424表达通过引起E3泛素连接酶支架蛋白cullin-2的转录后抑制而减弱HIF-1α的降解。
然而,在PASMCs中,粘附分子CD146和HIF-1α之间通过NF-κB通路的交叉调节已被证明可触发肺血管重塑。PASMC中CD146/HIF-1α轴的破坏会减缓血管重塑,并产生显著的PH值衰减。此外,PASMC过度增殖发生在暴露于生长因子(如表皮生长因子(EGF)、FGF2、PDGF),或者由HIF-1α介导,而不是由HIF-2α激活介导。这表明HIF-1α在这些生长因子的下游发挥作用。这些生长因子在PH中被公认为疾病驱动因子。PPARγ激动剂通过PPARγ介导抑制HIF-1α及其下游基因(如PDK-1、TRPC1和TRPC6),从而对PASMC发挥抗增殖作用。然而,缺氧通过HIF-1α在PASMC中诱导PPARγ下调,提示PPARγ和HIF-1α之间存在负反馈环机制。在其他调节因子中,缺氧诱导的miR-206和miR-150下调,通过靶向HIF-1α促进PASMC的促增殖和促迁移表型。此外,在大鼠PASMC中,SUMO-1和去磺酰化(SENP1)相关分子的上调会增加HIF-1α稳定性和转录活性,从而促进增殖。
线粒体异常
线粒体异常、NADPH氧化酶(NOX)和ROS之间的相互作用已被确定为PH肺血管细胞中HIF-1α的重要激活因子。例如,线粒体异常将代谢从氧化磷酸化转移到糖酵解(尤其是丙酮酸脱氢酶激酶[PDK]激活)导致电子通量的常氧损伤和线粒体ROS生成减少。这种假缺氧信号与HIF-1α的核移位有关。相反,报告表明,在IPAH患者的PAEC中观察到的超氧化物歧化酶2(SOD2)水平较低,由于ROS水平升高,可促进HIF-1α的稳定。由于肺血流动力学应激对PASMC施加机械牵拉,导致线粒体复合物III介导的ROS形成,其既诱导NF-κB通路又抑制Phd2,导致HIF-1α激活,表明血流动力学应激本身作为HIF-1α的独立调节因子。事实上,作为ROS上游调节器的几个分子与HIF-1α激活有关。例如,sirtuin 3是PASMC线粒体功能的关键调节因子,它的缺失导致线粒体功能障碍,导致ROS的产生和HIF-1α的稳定。因此,Sirtuin3敲除小鼠出现自发性PH和RV肥大。最近,我们发现了一种分子机制,支架蛋白,Ras结合域家族1A(RASSF1A)是PASMC中HIF-1α信号传导的关键调节因子。缺氧时,HIF-1α上调RASSF1A的表达,RASSF1A通过ROS驱动和蛋白激酶C介导(PKC介导)的磷酸化而稳定。RASSF1A反过来稳定HIF-1α,导致HIF-1转录活性增加。这个关键的RASSF1A–HIF-1α前反馈环路决定了PASMC和肺动脉外膜成纤维细胞(PAAFs)的增殖和糖酵解转换。通过基因消融RASSF1A破坏RASSF1A/HIF-1α的干扰,可减轻慢性缺氧小鼠肺血管重塑。
在肺血管细胞长期暴露于低氧水平的情况下,HIF亚型转录激活一系列调节血管张力、血管生成、代谢和增殖的基因(图2和图3)。此外,最近的研究强调了HIFs的潜在作用,以及其在PH相关的先天性和适应性免疫系统失调中潜在的分子机制。
PAECs(肺动脉内皮细胞)
PAEC在PH发病过程中表现出不同的表型(增殖、迁移、血管生成和/或内皮-间充质转化[EndoMT]),HIF亚型在定义这些表型中起着决定性作用。例如,HIF-1诱导细胞周期蛋白依赖性激酶抑制剂1B(p27Kip1)上调和细胞周期蛋白D1下调,导致缺氧PAEC的增殖和迁移减少。相反,HIF-2驱动的八聚体结合转录因子4(OCT4)表达,通过miR-130/131介导的PPARγ/apelin信号下调,导致PAEC增殖增加。此外,PAEC中的HIF-1和HIF-2都通过调节不同线粒体酶的表达来改变代谢表型,如通过调节丙酮酸脱氢酶激酶1(PDK1)、己糖激酶1,2(HK1,2)、乳酸脱氢酶A(LDHA)和葡萄糖转运蛋白1,3(GLUT1,3)以调节厌氧糖酵解和Warburg效应(有氧糖酵解)。HIF-1对糖酵解代谢的影响已得到充分证实;IPAH中的Warburg效应可能由HIF-1α稳定驱动,与缺氧环境无关。另一方面,HIF-2,而不是HIF-1,通过上调SNAI1转录因子,触发EndoMT,这一机制可能参与IPAH中闭塞性内膜/新生内膜病变和严重的肺血管壁增厚的发展。此外,EC HIF-2通过精氨酸酶-1依赖性机制影响缺氧PH的发展。HIF-2/精氨酸酶-1轴使血管NO稳态失调,导致缺氧小鼠的PH。因此,EC精氨酸酶-1的缺失可减轻缺氧小鼠的PH,而精氨酸酶抑制可组织野百合碱(MCT)小鼠的PH。然而,HIF-2介导的血管生成素-1和-2在内皮细胞中的表达对于维持适当的肺血管内稳态至关重要。这些数据表明HIF-1和HIF-2通过调节PAEC中不同的细胞过程在PH中发挥致病作用。
本篇文章来源于微信公众号: CardiothoracicSurgery