Soni Savai Pullamsetti, et al. Hypoxia-inducible factor signaling in pulmonary hypertension. The Journal of Clinical Investigation 2020


在肺血管细胞长期暴露于低氧水平的情况下,HIF亚型转录激活一系列调节血管张力、血管生成、代谢和增殖的基因(图2和图3)。此外,最近的研究强调了HIFs的潜在作用,以及其在PH相关的先天性和适应性免疫系统失调中潜在的分子机制。
PAECs(肺动脉内皮细胞)
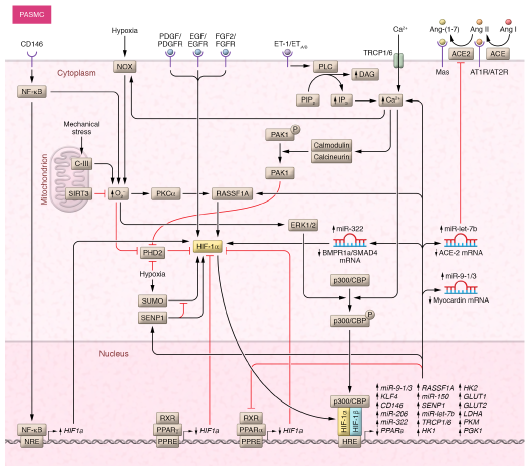
PAEC在PH发病过程中表现出不同的表型(增殖、迁移、血管生成和/或内皮-间充质转化[EndoMT]),HIF亚型在定义这些表型中起着决定性作用。例如,HIF-1诱导细胞周期蛋白依赖性激酶抑制剂1B(p27Kip1)上调和细胞周期蛋白D1下调,导致缺氧PAEC的增殖和迁移减少。相反,HIF-2驱动的八聚体结合转录因子4(OCT4)表达,通过miR-130/131介导的PPARγ/apelin信号下调,导致PAEC增殖增加。此外,PAEC中的HIF-1和HIF-2都通过调节不同线粒体酶的表达来改变代谢表型,如通过调节丙酮酸脱氢酶激酶1(PDK1)、己糖激酶1,2(HK1,2)、乳酸脱氢酶A(LDHA)和葡萄糖转运蛋白1,3(GLUT1,3)以调节厌氧糖酵解和Warburg效应(有氧糖酵解)。HIF-1对糖酵解代谢的影响已得到充分证实;IPAH中的Warburg效应可能由HIF-1α稳定驱动,与缺氧环境无关。另一方面,HIF-2,而不是HIF-1,通过上调SNAI1转录因子,触发EndoMT,这一机制可能参与IPAH中闭塞性内膜/新生内膜病变和严重的肺血管壁增厚的发展。此外,EC HIF-2通过精氨酸酶-1依赖性机制影响缺氧PH的发展。HIF-2/精氨酸酶-1轴使血管NO稳态失调,导致缺氧小鼠的PH。因此,EC精氨酸酶-1的缺失可减轻缺氧小鼠的PH,而精氨酸酶抑制可组织野百合碱(MCT)小鼠的PH。然而,HIF-2介导的血管生成素-1和-2在内皮细胞中的表达对于维持适当的肺血管内稳态至关重要。这些数据表明HIF-1和HIF-2通过调节PAEC中不同的细胞过程在PH中发挥致病作用。
PASMCs(肺动脉平滑肌细胞)
在PASMC中,HIF亚型不仅调节与细胞增殖和合成表型相关的基因,还调节与血管收缩(Ca2+调节/离子通道)、氧化应激、线粒体断裂和肾素-血管紧张素-醛固酮系统相关的基因(图3)。缺氧条件下观察到的PASMC增殖增加和合成表型转换是由HIF-1驱动的miR-9-1和miR-9-3的表达介导的,miR-9-1和miR-9-3负向调节心肌蛋白(myocardin,MYOCD)的表达。大鼠PASMCs增殖增强与BMP通路抑制有关,这是HIF-1而非HIF-2诱导miR- 322的结果, miR- 322导致Bmpr1a和Smad5基因的翻译后抑制。HIF-1依赖的miR-210上调通过靶向转录因子E2F3引起PASMC的凋亡抵抗。相反,HIF-2通过上调血栓反应蛋白-1促进缺氧反应性PASMC的迁移和收缩。因此,多种机制有助于PASMC的促增殖、促迁移和抗凋亡表型。此外,缺氧诱导的非肌化肺小动脉的肌化,包括本就存在的平滑肌细胞(SMC)祖细胞,这些祖细胞经历去分化、向远端血管迁移、增殖和再分化。Sheikh等人的研究表明,这些祖细胞的激活始于HIF-1α介导的PDGF-β表达,并通过HIF-1α介导的Krüppel样因子4(KLF4)表达,随着祖细胞来源的平滑肌细胞的扩增而进展。这些研究表明HIF-1在缺氧诱导的PH中肺动脉肌化的起始和进展中起着核心作用。细胞内K+和Ca2+浓度([Ca2+]i)的变化在调节PASMC的收缩、迁移和增殖中起着关键作用。值得注意的是,HIF-1通过调节各种离子通道的表达,在调控PASMC中[Ca2+]i水平方面起着重要作用。HIF-1促进瞬时受体电位(TRPC)通道成员TRPC1和TRPC6的过表达,随后增加缺氧PASMC中的[Ca2+]i。此外,HIF-1通过ET-1抑制电压门控K+通道成员,随后也增加[Ca2+]i。另一方面,HIF-1激活钙敏感K+通道BKCa的β1亚基(KCNMB1)的表达,从而阻止PASMC中[Ca2+]i过度升高。

PAAFs(肺动脉成纤维细胞)
虽然缺氧时PAAF中HIF-1α和HIF-2α都被激活,但HIF-2α诱导似乎在增殖反应中起主导作用,而HIF-1和HIF-2都以相似的程度增加PAAF的迁移。此外,研究表明HIF-1通过调节ACE和ACE2(ACE的同源物,可平衡ACE的功能)直接参与肾素-血管紧张素-醛固酮系统的调节,从而促进PAAF的增殖。
炎症细胞
免疫细胞通过调节肺血管细胞的功能在肺血管重塑中发挥重要作用。例如,缺氧的PAAFs在HIF-1的控制下驱动促纤维化巨噬细胞表型,导致各种旁分泌因子的释放。重要的是,巨噬细胞产生的VEGF和IL-6可促进肺血管重塑。
考虑到HIF通路的多种细胞和机械作用,HIF通路的基因敲除小鼠模型(Hif1/2α、VHL和Phd2)为HIF通路在肺血管系统缺氧适应和PH发展中的作用提供了许多有价值的见解(表2)。早期的研究表明,HIF通路的基因对胚胎发育至关重要,而这些基因中大多数双等位基因缺失是致命的。例如,小鼠中Phd2的纯合缺失会导致胚胎致死,而Phd1/3双敲除小鼠可以存活和繁殖。由于各种发育缺陷,小鼠中Hif1a、Hif2a或Hif1的完全缺失导致胚胎死亡。相反,Hif1a或Hif2a杂合缺失的小鼠进入成年期,在稳态条件下不显示表型,这使得它们有助于研究HIF在疾病中的作用。例如,Hif1a杂合缺失的小鼠在缺氧暴露时表现出PH和RV肥大减轻。相反,Hif2a杂合缺失的小鼠完全不受缺氧诱导的PH的影响。此外,为了评估成年期HIF亚型的整体缺失如何影响缺氧诱导的PH,Hu等人表明,小鼠的整体Hif1a缺失并不能阻止缺氧诱导的PH,而Hif2a整体缺失的小鼠在长期缺氧下无法存活。相反,在成年小鼠中,Hif2a部分缺失可减缓5周时缺氧诱导的PH的发展。此外,为了阐明HIF通路组分在PH发病机制中的细胞特异性和出生后特异性作用,各种研究使用了组成性或诱导性细胞特异性敲除小鼠模型。例如,Ball等人报告说,SMC特异性的Hif1a产后缺失(诱导性)可减轻PH,但不影响RV肥大。同时,在另一项研究中,SMC特异性的Hif1a缺失(组成性)小鼠表现出加重缺氧诱导的PH。最近的研究表明,EC特异性的Hif1a缺失(组成性)小鼠不能避免缺氧诱导的PH和RV肥大。然而,在另一项研究中,EC Hif1a缺失(诱导性)小鼠在缺氧条件下可免受PH和RV肥大的影响。有趣的是,内皮细胞和平滑肌细胞中诱导的Hif1a缺失并不能阻止缺氧诱导的PH和RV肥大。相反,在缺氧小鼠中,(PDGFR-β+)/SMC标记+祖细胞的产后缺失可完全阻止PH和RV肥大。关于HIF-2α,Skuli等人报告说,由于血管漏入肺实质,具有EC特异性Hif2a缺失的小鼠出现PH和RV扩张(但不是RV肥大)。类似地,Tang等人证明了Hif2a的EC特异性缺失,而不是Hif1a,可以阻止小鼠在缺氧条件下产生PH。有趣的是,内皮细胞中同时缺失Hif1a和Hif2a也可以防止博莱霉素诱导的PH和RV肥大,尽管肺纤维化正在发展。这些数据表明,Hif2a在内皮细胞中的显著作用在PH的启动和进展中至关重要。然而,EC特异性基因缺失小鼠模型的结果应谨慎解释,因为根据使用的是Cre/ERT2还是Cre23系统,基因缺失可能仅是EC特异性的,或者分别针对其他细胞类型。
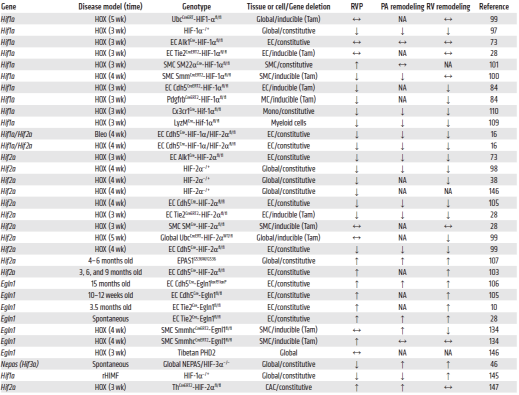
值得注意的是,多组研究表明,ECs中Phd2缺失的小鼠自发出现严重PH,并伴有大量肺血管病变和不良RV重塑,这在1.5个月大的时候就很明显了。ECs 中 Phd2 和 Hif1a 或 Phd2 和 Hif2a 的同时缺失表明,HIF-2α激活是PH发展中PHD2 缺乏的关键下游调节因子。有趣的是,这些小鼠在6-9个月龄内死亡率增加,可能是由于进行性RV衰竭。具有杂合和纯合 Hif2a G536W 功能获得性突变的小鼠会发生自发性 PH 和 RV 肥大,而没有 RV 扩张,这支持了HIF-2α活性在PHD2缺陷效应中的作用。此外,携带人类R200W VHL突变纯合子的小鼠(如在Chuvash红细胞增多症患者中发现的一样)出现PH。该模型中PH的发展在HIF-2α杂合缺失的情况下减弱,但在HIF-1α杂合缺失的情况下没有减弱,这表明HIF-2α在VHL功能丧失诱导的PH中起显著作用。此外,在一项探索HIF亚型在PH发病机制中的炎症和免疫特异性作用的研究中,EC特异性缺失PHD2的小鼠由于Hif2a的稳定而出现自发性PH,可通过移植WT骨髓来源细胞的而部分减弱,这提示了骨髓来源细胞中HIF-2的激活有助于肺血管重塑。此外,髓系细胞中Hif1a缺失的小鼠可部分避免低氧诱导的PH和RV肥大,这主要是由于巨噬细胞活性减弱所致。同样,单核细胞特异性Hif1a缺失的小鼠在缺氧或缺氧加Sugen 5416(SuHx)暴露下表现出PH、PA重塑和RV肥大的减轻。除髓系细胞外,HIF通路还参与淋巴细胞的调节。例如,Phd2缺失诱导的Hif2a激活导致免疫调节功能障碍,这可能部分解释了与调节性T细胞功能降低相关的PH发展。总之,这些研究表明,骨髓来源的巨噬细胞和胸腺来源的T细胞都参与肺血管重塑,至少部分原因是因为HIF通路的激活。综上所述,HIF亚型和HIF通路分子的整体、可诱导性和细胞特异性缺失,在成年小鼠PH发育的早期和晚期中确定了HIF亚型的细胞类型和环境特异性作用。
RV衰竭是PAH最常见的发病和死亡原因之一。PH开始时,右心室发生重塑以维持其收缩性,其特征是心肌细胞肥大和细胞外基质沉积介导右心室壁厚度和质量增加,适度扩张。然而,在持续压力过载过程中的某一时刻,RV的补偿机制失效,RV失效。在生理条件下,HIF-1α在右心室的表达明显高于左心室。RV HIF-1α在许多PH动物模型中表达增加,包括MCT大鼠、缺氧暴露(HOX)大鼠、肺动脉环缩(PAB)大鼠、SuHx大鼠和肺栓塞(PE)大鼠。PE大鼠RV HIF-1α表达增加,其水平与RV肥大和PAP呈正相关。有趣的是,具有Hif2a功能获得性突变的小鼠出现RV肥大,但尽管PAP显著增加,也未显示RV扩张的迹象,表明RV功能保留。在矫治后的法洛四联症患者中,由于TGF-β1(TGFB1)表达增加和心肌纤维化,HIF-1α功能获得性突变的存在与RV功能的保留和更好的预后相关。总的来说,这些报告表明,两种HIF的轻度至中度激活可能与RV功能的保留有关。相反,HIF的强烈激活会对RV功能产生不利影响。
例如,由于严重的红细胞增多症和扩张性心肌病,Phd2基因缺失的小鼠死亡率增加。类似地,在EC2特异性Phd2缺失的小鼠中激活HIF-2会导致自发性PH,严重RV衰竭导致高死亡率。然而,在RV肥大和衰竭的固定后负荷模型中,尚未使用细胞特异性基因敲除或过表达研究HIF和PHD的作用。
如上所述,在啮齿动物模型上的实验表明,HIF-1/2α对肺血管重塑具有深远的影响。此外,针对Hif2(而非Hif1)的反义寡核苷酸减少了暴露于缺氧的小鼠的血管肌化、PAPs升高和RV肥大,这表明抑制HIF-2α可以提供一种治疗方法来预防或逆转PH的发展。因此,人们对开发针对这一途径的疗法产生了极大的兴趣。在各种PH啮齿动物模型中,研究特别针对HIF通路的成分,如PHD2、HIF-1α或HIF-2α,并使用药理学药物。大多数直接或间接抑制HIF的受试药物都能够预防或逆转实验性PH(表3)。已评估了在mRNA水平(拓扑替康和喜树碱)、蛋白质合成(2-甲氧基雌二醇、地高辛、塞拉霉素、咖啡酸苯乙酯)、蛋白质积累和转录活性(YC-1)以及靶向调节HIF轴的分子(抗CD146、单克隆抗体AA98、芹菜素),或在mRNA水平(C76)或在异源二聚化和DNA结合水平(PT2567)抑制HIF-2α的药物/化合物。这些通过不同途径(腹腔内、静脉内、皮下、口服)给予的抑制剂在各种PH动物模型(缺氧、MCT和SuHx)中显示可以预防和逆转PH。值得注意的是,HIF-2α抑制剂C76在三种PH的实验模型中显示出强烈的抗重塑作用,表明抑制HIF-2可能是治疗PH的一种有希望的方法。
本篇文章来源于微信公众号: CardiothoracicSurgery